
Arc Virtual Cell Challenge part 3: how to train your own Virtual Cell Model
Let's go through the example notebook on running STATE, Arc's Virtual Cell model.

Here is my third article covering the Arc Virtual Cell Challenge (see first and second).
Right at the beginning of the challenge, the organizers released a Colab Notebook to help participants get started. This notebook shows how to train STATE, the Virtual Cell Model from the Arc Institute, to predict the effect of perturbations in H1 cell lines.
The notebook is well written and it is self-explanatory, and I’ve read it many times. Let me guide you through it.
What is STATE, and how does it work?
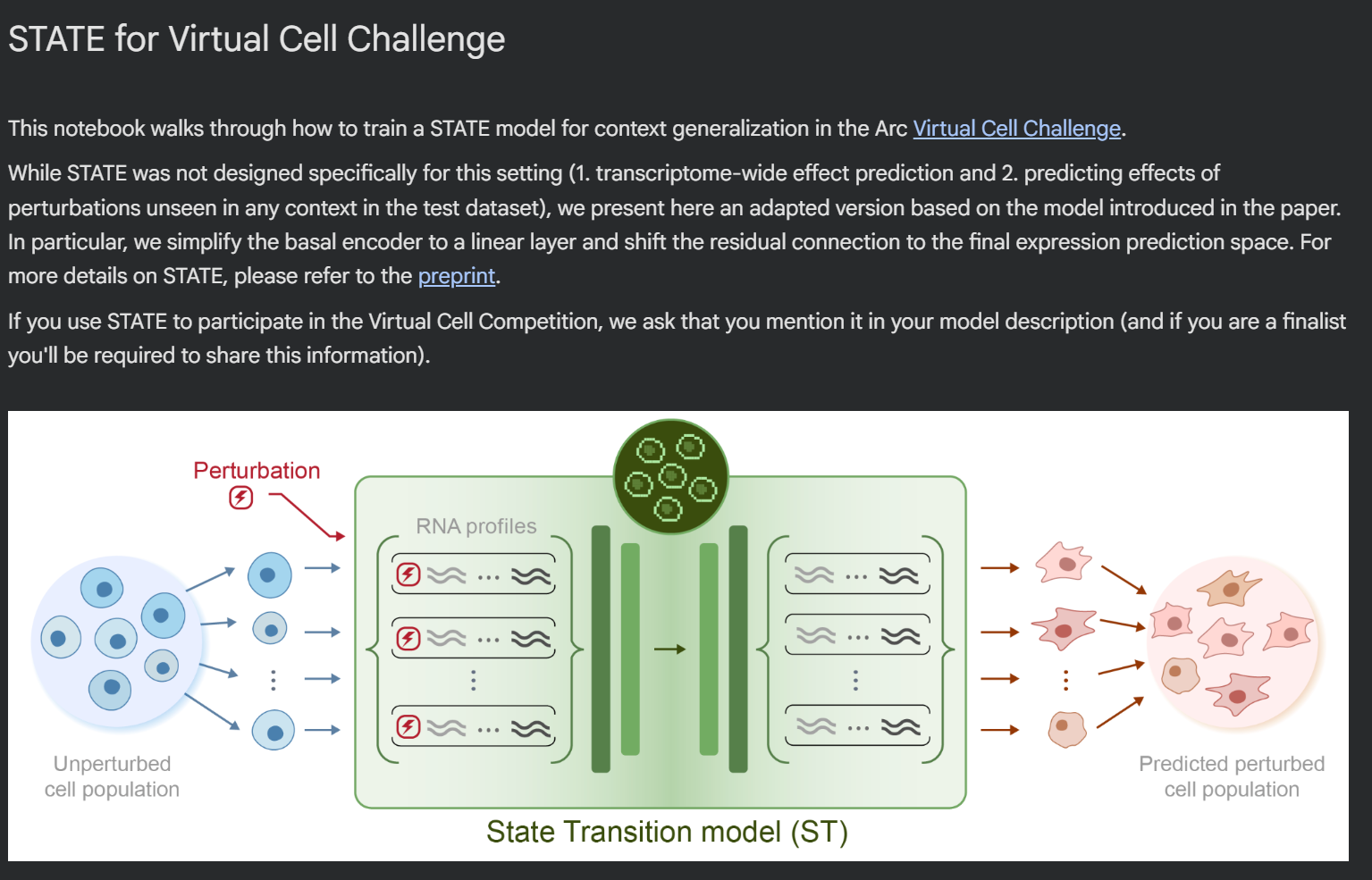
STATE is the Virtual Cell Model from the Arc Institute.
It is composed by two modules: State Embedding (SE) and State Transition (ST).
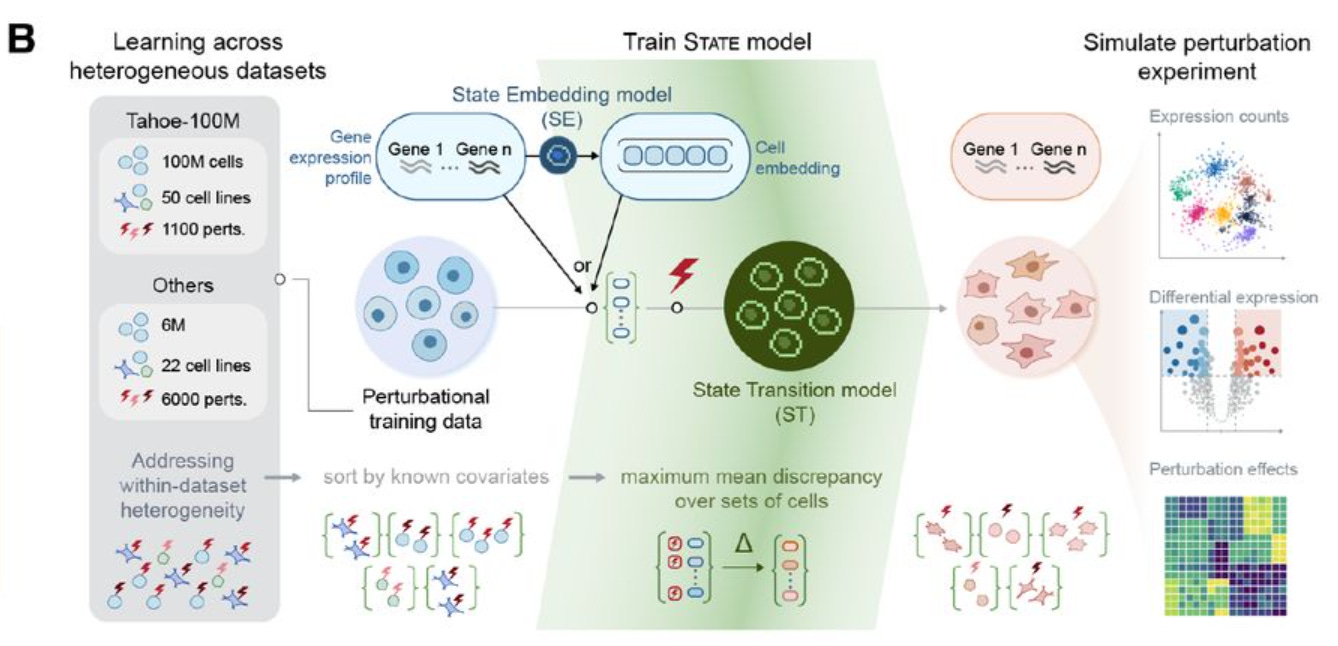
The first module, SE, is an encoder that takes a single-cell transcriptome as input, and transforms it into vectors of numbers (embeddings), that can be used as input to other learning tasks. These embeddings are corrected for technical artifacts (batch effects), and can learn biological interactions - for example, cells from the same cell line will cluster together in the vector space. We don’t use SE directly in the notebook, but potentially these embeddings could be used for other machine learning approaches.
The second module, ST, is a transformer that can predict how cells will transition in response to a perturbation. It is trained on a dataset containing both control cells (where no gene has been perturbed) and perturbed cells; and its output, when used for inference, is a AnnData file containing simulated gene expression of cells exposed to perturbations.
How to use STATE to predict the effect of gene perturbations?
As explained in a previous post and in the notebook itself, the idea is to use STATE to predict the effect of perturbations in the H1 cell line, using data from the public domain.
This is a two-step regression problem. First, we need to learn the effect of each specific gene perturbation. We can learn it by looking at what happens in other cell lines when the same genes are silenced - there are a few well known datasets available with that information, notably the Replogle and the Nadig ones.
The second problem to solve is the cell line context. The H1 cell line is from stem cells, while the cell lines in the Replogle and Nadig datasets are from cancer cell lines; so, we there may be biological mechanisms specific to H1. To learn about these differences, we can use the training dataset provided by the competition, which contains the output of a number of gene perturbations in this cell line.

As mentioned earlier, Virtual Cell Models like STATE can in principle handle this two-step problems, where we want to predict the effect of perturbation in a different context.
Three Engineering things I’ve learned from this notebook
There are at least three clever things that I have learned from this notebook, in terms of engineering.
The first is the use of uv for managing dependencies. uv is a new tool to manage python environments - a replacement for conda, pip, and the likes. It is also very, very fast - is there anything better than seeing python dependencies being installed in a matter of seconds?
The second thing I liked is the use of Weight and Biases (wandb) for logging the runs. I am used to ML Flow, but I have to say that wandb’s interface is much better - more clean and easy to use. It makes it easier to handle many runs and summarize the results.
The third thing is the clever use of .toml files to manage configuration for the training. The state github repository contains some examples of configuration files. These config file work together with other .yaml files in the state folder. It took me a while to understand the config structure, but in the end, I found it very flexible and powerful.
How is the training of STATE evaluated?
For me, this was the first time I trained a model of this size from scratch. I used weights from HuggingFace, but not trained on new data.
So the question is, how to evaluate the performances of the training, to see if we are overfitting or not? How to do hyperparameter optimization? It would be very costly to do cross-validation, and it would take ages.
The answer is zero-shot and few-shots validation.
In zero-shot, we use an entire cell line as validation or test set. In the notebook they suggest to use the Hepg2 cell line as validation. Of course, zero-shot is difficult the model does not know anything about Hepg2 cells… It could be any random cell line, for what we know. We need to assume that Hepg2 cells are similar enough to the other lines used for training, in terms of their biology.
In few-shots, we use specific perturbations to evaluate performances. We need to choose a proper list of genes, keeping a mix of essential and non-essential genes, covering a mix of biological functions, to use as validation for our model. These perturbations will be kept out of the training data, and used to measure how good the model is predicting.
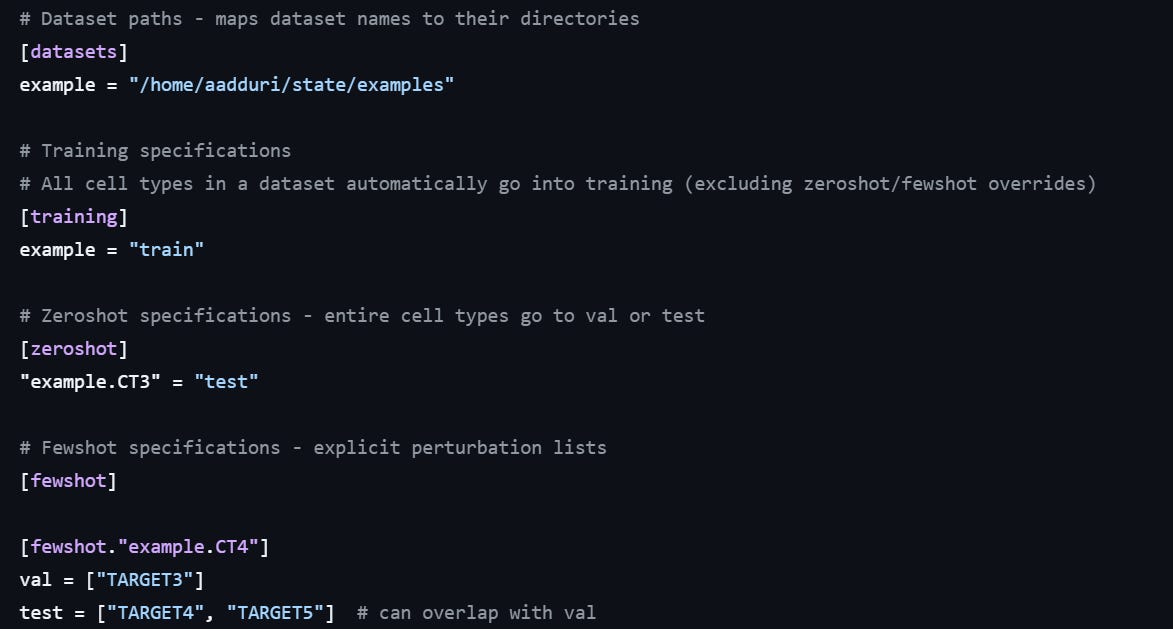
The config file above shows how this is set up in STATE. In this example, the “CT3” cell line will be used as zero-shot, while TARGET3, TARGET4 and TARGET5 will be used as validation and test.
How difficult is it to train STATE, in practice?
The google/colab shows how to trigger the training of STATE, assuming data and configuration is in order:
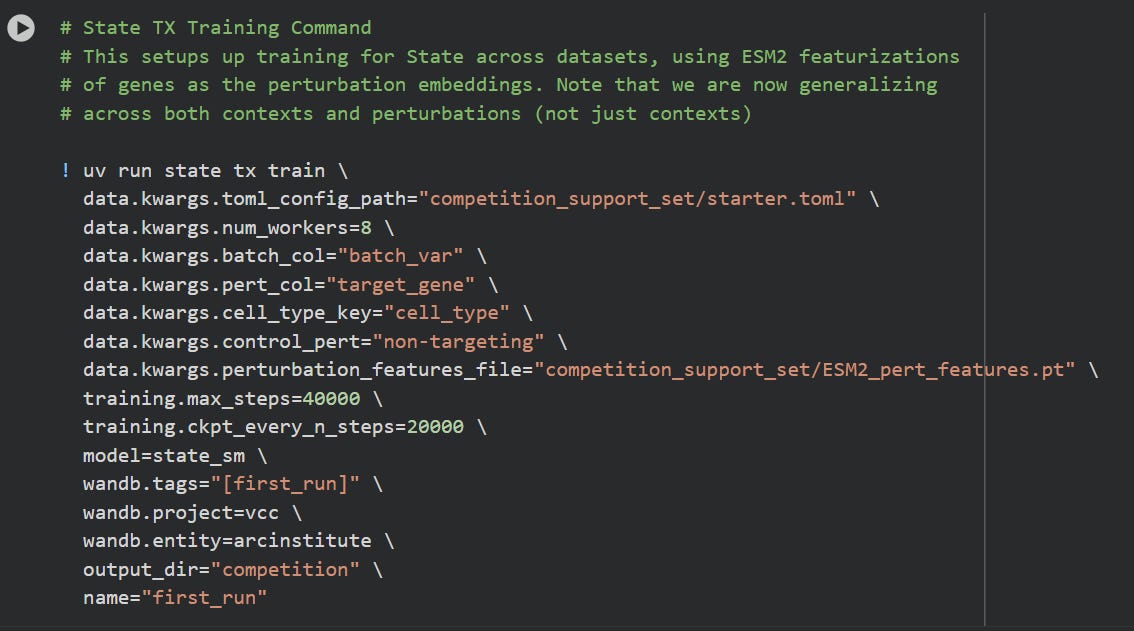
If you let this run, this will take a loooong while to finish. It will likely crash, on google colab.
One challenge for me was to find compute resources for this - I am grateful to mithril.ai for the free credits, and to Kiin Bio for some amazon support.
At the beginning, we struggled to train the model, because it just never converged. Here is a screenshot of a 20K epochs training run, also known as 7-8$ of GPU burned:
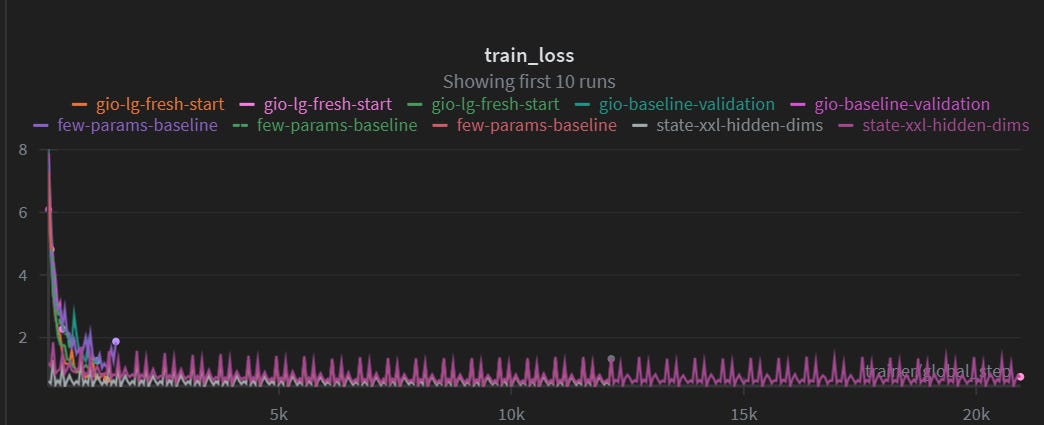
What can be done to improve the predictions?
What can be done to improve the training of STATE?
One approach is to fork the STATE code from github, make changes to the architecture, and see what works. For example, use a different base transformer than LlamaBidirectionalModel, change the number of layers, and play with different architectures.
Another approach is to add more data, and choose carefully what to include in training set or validation. There are not many other large-scale perturbation datasets in cell lines, unfortunately - the biggest dataset, TAHOE-100M, unfortunately is based on drug pertubations instead of gene knock-outs. But there is space to integrate biological knowledge in the training, or at least to refine the predictions.
I believe a lot of information on this will come out once the challenge finishes, and the winner announced. That’s going to happen soon, in a few weeks.
My articles on the Arc Virtual Cell Challenge
Thanks for reading my article!
In this post, we learned how to train the STATE Virtual Cell Model, using the notebook provided by the organizers.
Here are my other articles on this series:
Arc Virtual Cell Challenge part 1: my diary

The Arc Virtual Cell Challenge is a competition organized by the Arc Institute in the summer of 2025. The challenge is still ongoing, with the final deadline on November 17.
Arc Virtual Cell Challenge part 2: What the Leaderboard Collapse Reveals About Model Evaluation

The Arc Virtual Challenge is coming to an end, with the deadline in a mid November 2025.
